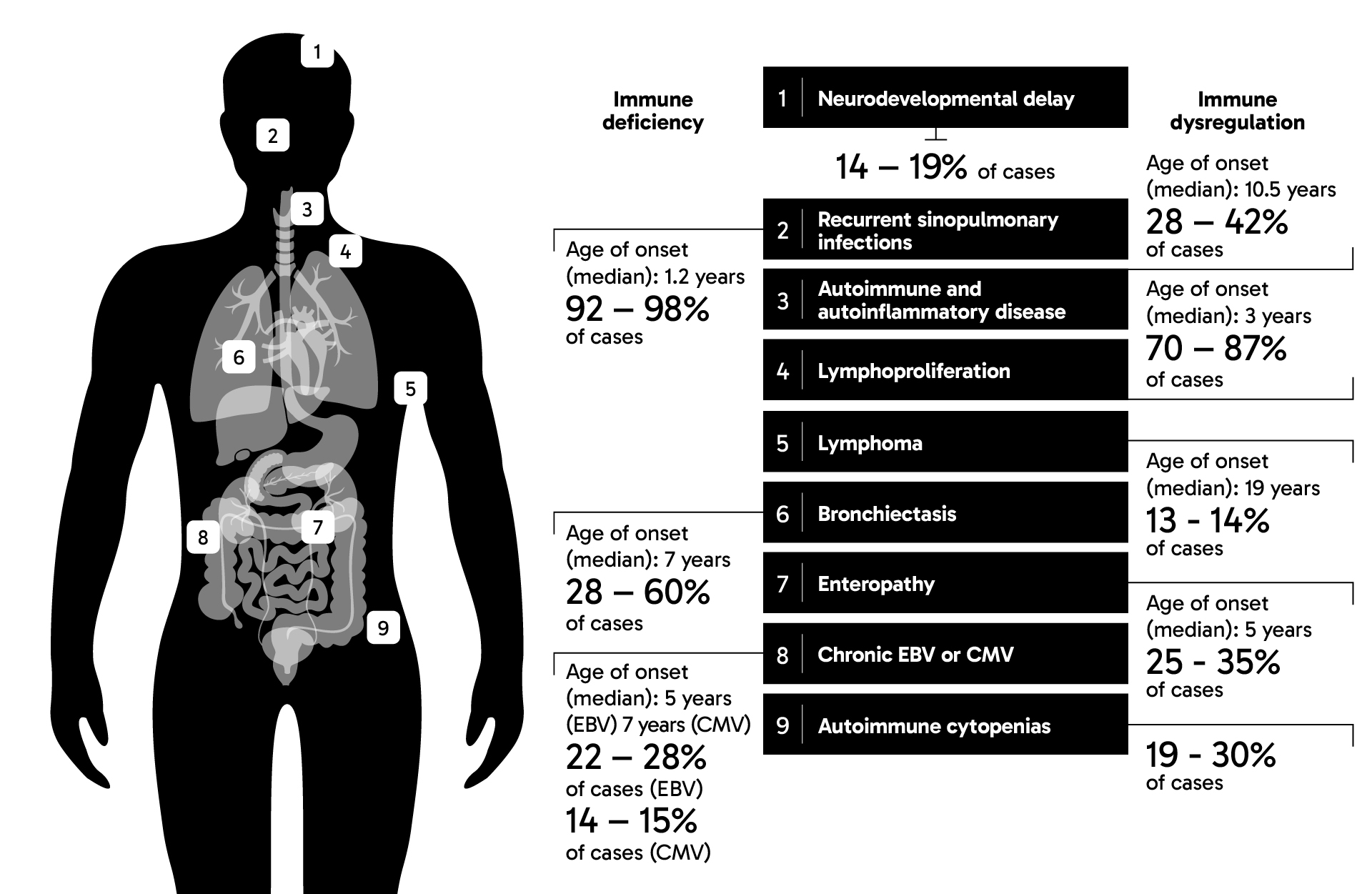
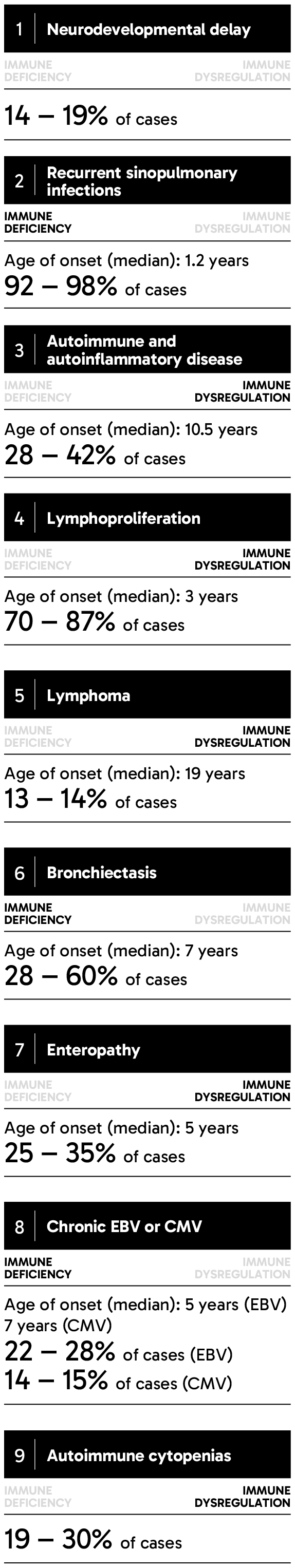
APDS can be inherited in an autosomal dominant pattern, meaning that a person can inherit a variant gene from only one parent for the disease to manifest.3 Children born to a parent with variants in either one of two genes encoding phosphoinositide 3-kinase delta (PI3Kδ) will have a 50% chance of inheriting APDS.4
50%
CHANCE AN AFFECTED PARENT WILL PASS AN APDS GENE ON TO THEIR CHILD.