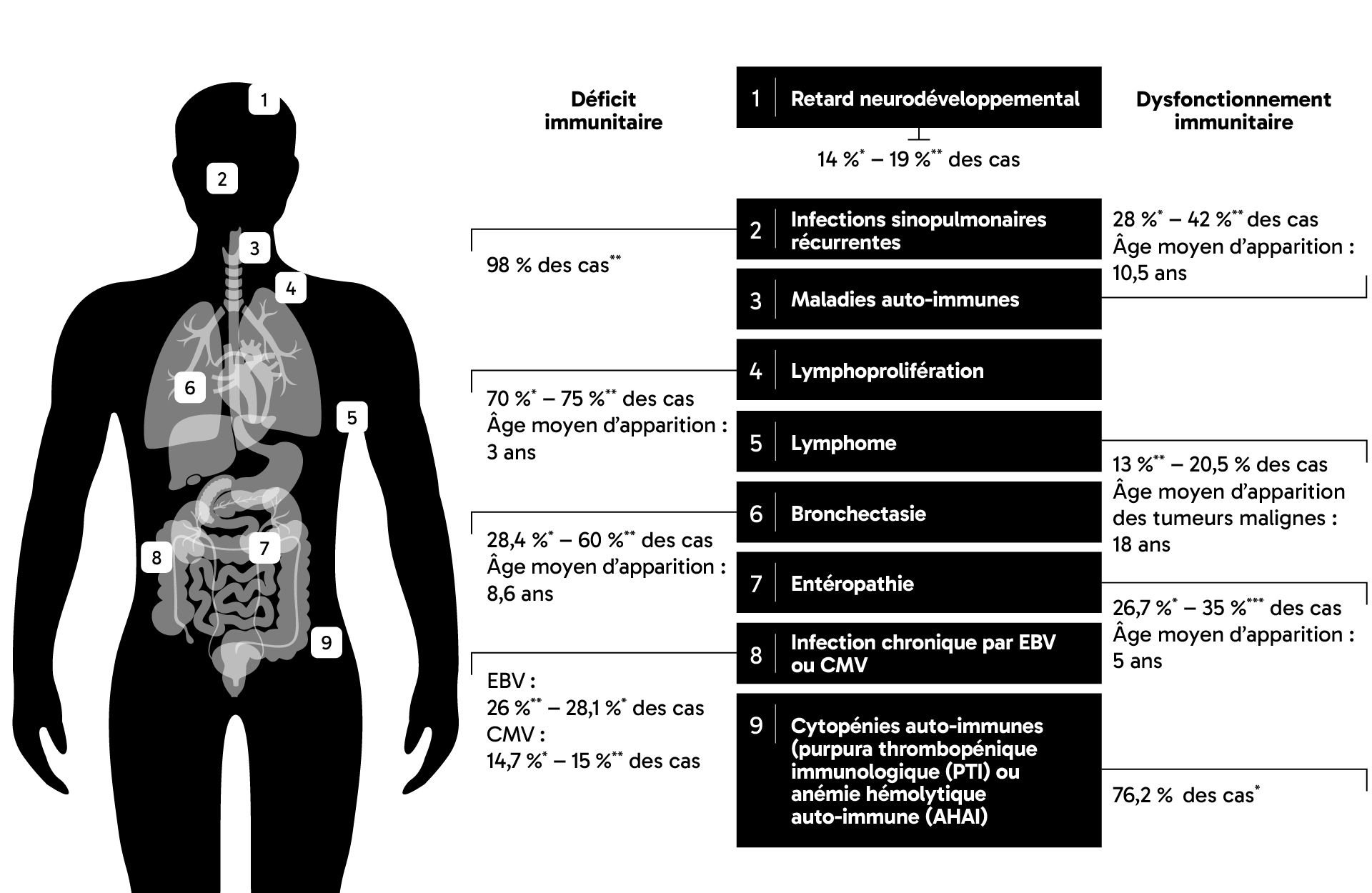
L’APDS se transmet sur un mode autosomique dominant par l’un des deux parents. Cela signifie qu’il suffit d’un seul gêne touché par l’anomalie génétique pour que le patient soit malade. Si un parent est porteur d’une mutation sur l’un des 2 gènes codants pour la PI3Kδ, le risque de transmission de la maladie à son enfant est de 50 %. 5,6
50 %
UN PARENT ATTEINT D’APDS A 50 % DE RISQUE DE TRANSMETTRE LA MUTATION DU GÈNE À SON ENFANT. 6