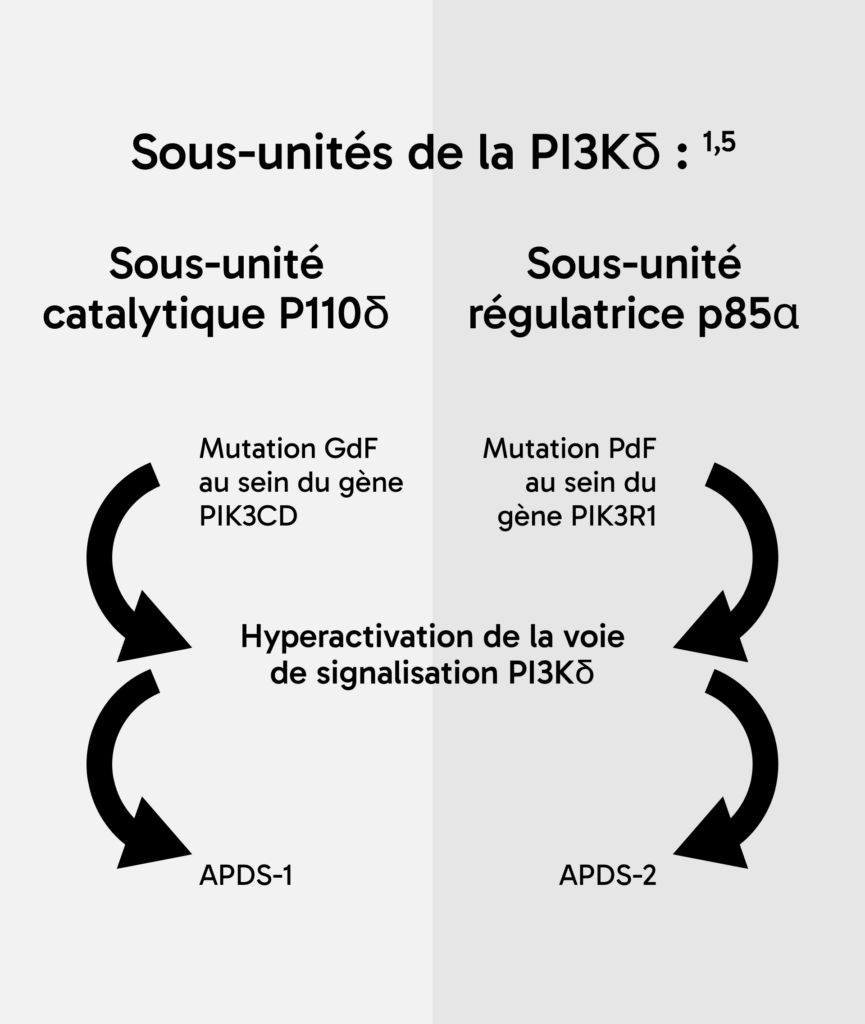
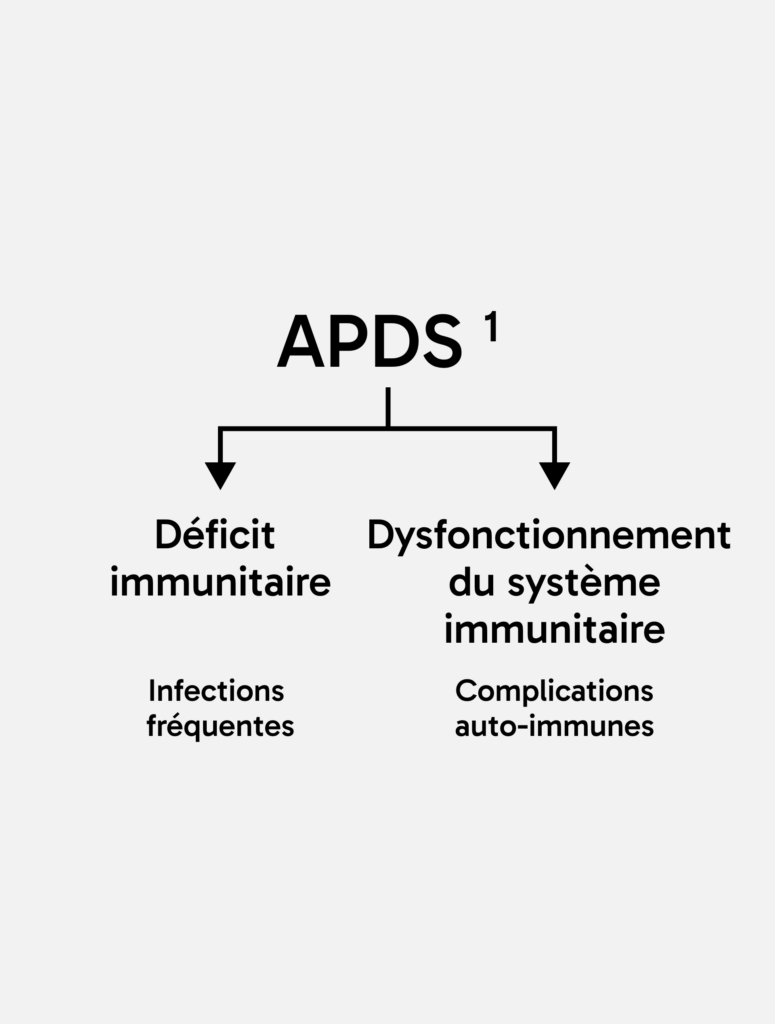
Une analyse de la littérature réalisée en 2024 a permis d’établir un taux de survie conditionnel sur 256 patients selon l’âge des patients atteints d’APDS : ce taux était de 87 % à 20 ans, de 74 % à 30 ans et de 68 % à l’âge de 40 et 50 ans***. 4
38,6 %
Une revue de la littérature a démontré que les antécédents familiaux immunitaires étaient présents chez plus d’un tiers des 243 patients atteints d’APDS (38,6 %)*. Cependant, l’APDS peut survenir spontanément, sans qu’aucun des parents ne soit porteur d’un variant génétique à l’origine de la maladie. On estime que l’APDS touche 1 à 2 personnes par million, adultes et enfants confondus. 5,6